Genetik:
Das Angelman-Syndrom wird durch fehlende Expression des UBE3A Gens im Gehirn verursacht. Der Chromosomenabschnitt 15q11-q13 auf Chromosom 15, auf dem dieses Gen liegt, unterliegt sogenanntem Imprinting. Das bedeutet, dass bestimmte Gene auf diesem Abschnitt ausschließlich auf dem vom Vater stammenden und andere nur auf dem von der Mutter stammenden Chromosom aktiv sind. Beim Angelman-Syndrom ist der mütterliche Chromosomenabschnitt nicht funktionstüchtig und das UBE3A Gen auf dem väterlichen Chromosom ist durch Imprinting stillgelegt; somit fehlt das Genprodukt komplett.
Ist nicht der mütterliche, sondern der väterliche Chromosomenabschnitt fehlerhaft, führt dies zum Prader-Willi-Syndrom.

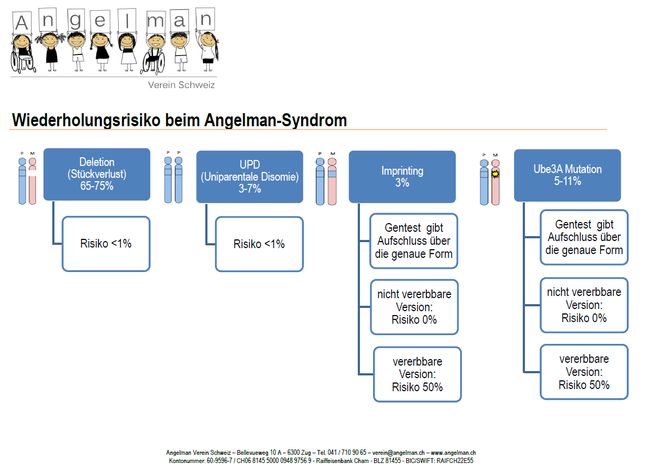
Bei 50 bis 80 von 100 Menschen mit Angelman-Syndrom liegt die Ursache der Besonderheit in einer Deletion (= Stückverlust) des mütterlichen (=maternalen) Chromosoms 15 im Bereich 15q11-q13 (zum Teil mit Translaktion).
Bei zwei bis fünf von 100 Personen liegt eine uniparentale Disomie (UPD) 15 vor. Im Fall des Angelman-Syndroms hat das Kind beide Chromosomen 15 vom väterlichen Elternteil geerbt (paternale Disomie) und keines von der Mutter.
Bei acht bis elf von 100 Menschen mit Angelman-Syndrom findet sich eine Genmutation im UBE3A Gen auf dem maternalen Chromosom.
Ein Fehler im Imprinting ist bei etwa fünf von 100 Personen nachweisbar. Dies kann aufgrund einer durch Epigenetik bedingten Mutation oder aufgrund einer Mutation oder Deletion des Imprinting-Centers der Fall sein. Dieses ist ein DNA-Abschnitt, der die differentielle epigenetische Modifizierung von sieben Genen auf dem paternalen und maternalen Chromosom kontrolliert. Dies führt jeweils zu eine Aktivierung oder Inaktivierung des Gens. Das Imprinting hat zur Folge, dass auf dem maternalen Chromosom das Gen UBE3A aktiv ist und exprimiert wird, UBE3A auf dem paternalen Chromosom jedoch inaktiviert ist. Ist die Funktion des Imprinting-Centers gestört, hat dies ein fehlendes oder fehlerhaftes Methylierungsmuster zur Folge, was unter anderem das Angelman-Syndrom zur Folge haben kann.
Das Angelman-Syndrom kann eine erbliche Komponente haben. Die Eltern sind dabei nicht betroffen, haben aber bestimmte Chromosomenbesonderheiten, die die Wahrscheinlichkeit erhöhen, ein Kind mit Angelman-Syndrom zu zeugen. Dies zeigt sich in der auffallenden Häufigkeit von gleichsam betroffenen Geschwistern. Eine Mutation im UBE3A Gen etwa kann über die männliche Seite einer Familie über Generationen still weitergegeben werden. Trägt der Grossvater eine Mutation, die er an seine Tochter weitergibt, macht diese sich auch in der Tochter noch nicht bemerkbar, da die Mutation sich auf dem väterlichen Chromosom befindet. Die Tochter hat dann aber ein Risiko von 50% ein Kind mit Angelman-Syndrom zur Welt zu bringen.
Diagnose:
Die Diagnose wird im Schnitt zwischen dem dritten und siebten Lebensjahr durch Neurologen (anhand auffälliger EEG-Werte, unabhängig von Epilepsie, auch in Schlaf bestehend) oder durch Genetiker (anhand einer zytogenetischen oder molekulargenetischen Untersuchung) gestellt.
Es ist möglich, das Angelman-Syndrom beim grössten Teil der betroffenen Kinder durch einen Gentest festzustellen. Bei einigen Betroffenen, bei denen man von der klinischen Symptomatik her zwar vom Vorliegen des Syndroms ausgeht, kann dies aber nicht nachgewiesen werden. Ein positiver Gentest kann also das Angelman-Syndrom mit Gewissheit feststellen, jedoch schliesst ein negativer Test es nicht aus.
Vielfach wenden sich die Eltern des Kindes mit dem Verdacht auf das Angelman-Syndrom an ihren Kinderarzt, da sie sich aufgrund bestimmter typischer Auffälligkeiten bereits im Vorfeld informiert haben, um eine Erklärung für Besonderheiten ihres Kindes zu finden. So kann die Beobachtung des Verhaltens und Aussehens des Kindes bei der Diagnostik eine wesentliche Hilfe sein.

Wiederholungsrisiko beim Angelman Syndrom